L’uso dei modelli matematici in biologia comporta una serie di condizioni e limitazioni. Un primo limite è la necessità di semplificare molto i modelli matematici, altrimenti la complessità di calcolo ne renderebbe inutile l’uso; il secondo è costituito dal fatto che i modelli descrivono solo una parte delle proprietà del sistema biologico. Spesso occorre limitarsi a «modelli empirici», relazioni ad hoc che descrivono il comportamento del sistema. In altri termini, non è possibile un atteggiamento riduzionista: le proprietà biologiche che si riscontrano a un certo livello di complessità di un sistema non sono semplicemente deducibili da quelle che governano il sistema ai livelli inferiori. Occorre quindi un approccio plurilivellare. In particolare è utile studiare il sistema biologico nel suo complesso senza scomporlo in pezzi: nell’ultima parte dell’articolo l’autore illustra i recenti aspetti di questo metodo nel campo di indagine che prende il nome di biologia dei sistemi.
Nella ricerca biologica è ormai pratica largamente diffusa l’utilizzo di modelli matematici. Dunque, quando si parla di approcci di questo tipo, è opportuno prima di tutto chiarire quali siano gli intenti degli sperimentatori nell’avvalersi di questi strumenti.
Come premessa, è bene precisare che modelli matematici applicati ai sistemi biologici possono essere empirici o meccanicistici. Quelli empirici sono generalmente basati su semplici osservazioni e descrivono il sistema senza una reale comprensione delle leggi che lo governano; vale il contrario per quelli meccanicistici, che si basano invece (o presumono di basarsi) sulla conoscenza effettiva delle leggi che governano i fenomeni investigati.
È evidente che i modelli meccanicistici sono quelli che maggiormente interessano, e in qualche senso attraggono, gli sperimentatori sia perché dovrebbero consentire predizioni più fedeli del comportamento del sistema, sia perché il loro successo è a sua volta diagnostico di una reale comprensione delle sue proprietà.
Infatti l’intento di ogni modellizzazione è la comprensione il più possibile approfondita delle proprietà del sistema e ultimamente, se possibile, la definizione degli aspetti più essenziali del fenomeno vita.
L’impostazione del problema
Introduciamo innanzitutto una sommaria descrizione del metodo e delle finalità con cui viene utilizzato lo strumento matematico per lo sviluppo di un modello meccanicistico: 1) si parte da una raccolta di dati sperimentali relativi a un oggetto (come una proteina o un sistema cellulare) e a partire da essi si sviluppano modelli, vale a dire equazioni che rappresentino le leggi chimico-fisiche che governano il sistema; 2) vengono successivamente effettuate misure sul sistema, in condizioni diverse, predefinite dallo sperimentatore, al fine di valutare se e in che misura il riscontro sperimentale si adatti alle predizioni del modello. Esso, in particolare, dovrebbe essere in grado di predire la riposta del sistema a variazioni ambientali, a partire dalle proprietà elementari di questo.
Dunque il metodo consiste di una parte sperimentale (raccolta di dati) e una teorica (rielaborazione per predire proprietà emergenti). Non c’è dubbio che in determinati casi questo approccio abbia funzionato (e quando funziona è sempre un evento felice). Ma, come regola, i successi sono normalmente parziali.
In linea di principio, l’applicazione di un modello a un determinato sistema può portare in due modi a una migliore comprensione del medesimo: 1) verificando se il modello matematico predica correttamente, o meno, il comportamento del sistema (e in entrambi i casi la comprensione del sistema ne risulta arricchita); 2) o anche ricavando i parametri quantitativi inerenti il modello, che sono quindi descrittivi delle proprietà del sistema (come per esempio le costanti di velocità nel caso di modelli di cinetica chimica).
Ciò detto, è anche opportuno precisare che, specie se applicati a sistemi biologici, i modelli presentano virtualmente in ogni caso due principali limitazioni, che si collocano a livelli metodologicamente distinti: 1) quasi sempre introducono delle semplificazioni più o meno sostanziali rispetto alle proprietà del sistema che dovrebbero rappresentare. Ciò è richiesto o dalla impossibilità di sviluppare equazioni che descrivano fedelmente il comportamento del sistema, oppure dalla complessità delle equazioni sviluppate, che le rende inutilizzabili sul piano pratico. 2) non meno importante, i modelli descrivono sempre una parte di tutte le proprietà del sistema. Tipico a questo riguardo è il fatto che esistono di norma sistemi descrittivi distinti per trattare le proprietà funzionali e le proprietà strutturali delle molecole proteiche, e come dato di fatto una integrazione completa dei due sistemi è con ogni evidenza ben lontana dall’essere attuabile.
I problemi legati alla complessità
Un’evidenza immediata dei problemi legati alla complessità e alla conseguente difficoltà di ricavare modelli che siano rigorosamente descrittivi del sistema, viene da semplici esempi tratti dalla cinetica chimica, anche di interesse della biologia (per esempio nell’ambito degli studi sul flusso metabolico).  Nel caso di una reazione irreversibile con un solo reagente e un solo prodotto, vale a dire:
Nel caso di una reazione irreversibile con un solo reagente e un solo prodotto, vale a dire:
l’equazione che dalla variazione del tempo della concentrazione di A è:
[A] = [A]0 e-kt
dove k è la costante di velocità tipica della reazione e t è il tempo. Si tratta di una tipica cinetica esponenziale inversa, come mostrato nell’immagine qui a sinistra.
Vediamo che in questo caso il modello matematico è molto semplice e facilmente manipolabile. Ma se si considera in aggiunta che la reazione sia reversibile, vale a dire:
l’equazione cinetica risulta la seguente:
![]()
 che corrisponde a un profilo nel tempo delle due specie chimiche illustrato nell’immagine a destra.
che corrisponde a un profilo nel tempo delle due specie chimiche illustrato nell’immagine a destra.
Dunque ciò che balza in grande evidenza è il fatto che una complessificazione apparentemente limitata del modello chimico già produce un’equazione enormemente più complessa.
Se infine consideriamo due reazioni consecutive irreversibili, vale a dire:
A -> B -> C
ciò da luogo alle seguenti equazioni cinetiche:

Ed al relativo grafico riportato nella seguente immagine:

Ciò che emerge con evidenza da questi esempi è che già in sistemi semplici, un modesto incremento di complessità si traduce in una notevolissimo incremento di complicazione dei modelli descrittivi. Non è difficile immaginare che la trattazione di casi anche solo leggermente più complicati produrrebbe equazioni inutilizzabili all’atto pratico. Da ciò emerge l’esigenza di semplificare i modelli perché possano essere praticamente applicabili ai casi concreti.
Un altro caso molto rappresentativo della complessità legata alla modellizzazione è quello dell’emoglobina, una proteina nota a tutti per il suo ruolo biologico consistente nel trasporto di ossigeno dai polmoni (o dalle branchie) ai tessuti periferici.  In base alle osservazioni sperimentali, il profilo di saturazione (vale a dire, la percentuale di molecole saturate dall’ossigeno) in funzione della pressione parziale di ossigeno (un parametro che esprime la concentrazione disciolta nel sangue) è data dal profilo nell’immagine a sinistra.
In base alle osservazioni sperimentali, il profilo di saturazione (vale a dire, la percentuale di molecole saturate dall’ossigeno) in funzione della pressione parziale di ossigeno (un parametro che esprime la concentrazione disciolta nel sangue) è data dal profilo nell’immagine a sinistra.
Si tratta dunque di un profilo di saturazione sigmoidale (caratteristico anche di molti enzimi e di altre proteine). Una tale risposta della molecola consente un’efficienza di trasporto dell’ossigeno ai tessuti molto maggiore di quanto non sia possibile in base ai classici profili iperbolici tipici degli enzimi. Infatti a livello dei distretti periferici la bassa pressione di ossigeno è associata con un rilascio di ossigeno maggiore di quello che si avrebbe se il profilo fosse iperbolico.
Ora, l’equazione a tutt’oggi utilizzata quasi sempre per descrivere questo profilo è stata sviluppata nel 1910 dal fisiologo Archibald Hill. Si tratta un modello semplicissimo ed empirico:

(Nell’equazione, θ indica la frazione di emoglobina legata all’ossigeno e pO2 la pressione parziale. Si noti l’esponente n, che conferisce l’andamento esponenziale)
È chiaro che questa equazione è puramente empirica e non meccanicistica; vale a dire non si basa sulla conoscenza del meccanismo di interazione ossigeno-emoglobina. Una equazione meccanicistica è stata sviluppata nella prima metà degli anni Sessanta del XX secolo da Jacques Monod e altri, ed è la seguente:

(in questo caso [S] sta ad indicare la pressione parziale di ossigeno).
È degno di nota il fatto che l’utilizzo di un modello empirico, a distanza di oltre un secolo dalla sua elaborazione, continui a prevalere su modelli meccanicistici molto più raffinati e incomparabilmente più realistici, proprio grazie alla sua semplicità. Ciò accade molto spesso nella modellizzazione di sistemi biologici in quanto, in vista di una descrizione delle proprietà del sistema investigato, si preferisce privilegiare la maneggevolezza del modello piuttosto che la sua rigorosa aderenza alle proprietà del sistema medesimo.
Solo un problema di complessità?
Gli esempi che abbiamo prodotto mostrano in modo molto evidente che nei sistemi biologici la modellizzazione si scontra con difficoltà considerevolissime già a livelli di complessità relativamente limitati. Ma la domanda che ci possiamo porre, sulla base di questa riflessione iniziale, è se tutti i problemi legati alla modellizzazione siano riconducibili a una insufficienza dello strumento matematico. Molti esempi suggeriscono che il problema sia invece più complesso.
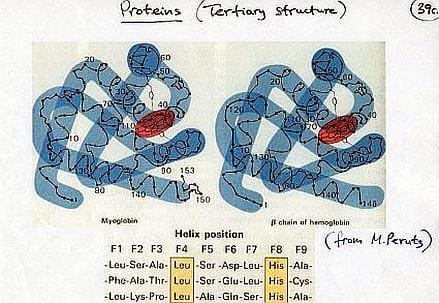
Possiamo documentare questo concetto prendendo in esame le proteine, macromolecole biologiche di primaria importanza (assieme agli acidi nucleici), che sostengono virtualmente ogni funzione biologica.
In termini di costituzione chimica si tratta di polimeri lineari derivanti dalla condensazione di un repertorio di 20 amminoacidi (nell’immagine seguente viene mostrata la modalità di associazione chimica degli amminoacidi e i diversi livelli strutturali delle proteine).
All’atto della sintesi esse sono filiformi, e già prima che la sintesi sia completata, o subito dopo, si raggomitolano raggiungendo una ben precisa struttura tridimensionale, detta nativa, cioè l’unica associata con la funzione che la proteina deve svolgere naturalmente. Oggi sappiamo con certezza che l’informazione che porta a questa struttura tridimensionale è contenuta tutta nella sequenza amminoacidica (quindi nella proteina stessa).
Tale quadro teorico di riferimento è ben consolidato e ha sollevato già all’avvio delle investigazioni sulle proteine (attorno agli anni Cinquanta-Sessanta del XX secolo) due domande fondamentali: 1) come possiamo predire la struttura dalla sequenza? 2) come possiamo predire la funzione dalla struttura?
Poiché la struttura che la molecola effettivamente assume è quella termodinamicamente più stabile (cioè con un minimo di energia), uno dei modi praticabili in linea di principio per predire la struttura dalla sequenza è computazionale: per esempio, partendo dalla conformazione più estesa della proteina, vengono calcolate conformazioni sempre più stabili che si formano a partire da quella estesa. In pratica, si simula in silico (cioè con il computer) il processo di protein folding, cioè il raggomitolamento della proteina che avviene entro le cellule a dare conformazioni sempre più stabili e compatte (molte simulazioni di questo tipo sono reperibili in Internet: si veda per esempio il sito: www.youtube.com/watch?v=meNEUTn9Atg).
In realtà, la potenza di calcolo che è richiesta per questa operazione richiede cluster di parecchi potenti computer che lavorino diversi giorni al fine di riprodurre un fenomeno che avviene sulla scala temporale di secondi o frazioni di secondo. Nonostante l’imponenza dei mezzi richiesti, questi metodi bioinformatici hanno una buona probabilità di successo solo con proteine la cui dimensione non ecceda i 100 amminoacidi o poco più (si tenga conto che una proteina di dimensioni medie consiste tipicamente di 300-400 amminoacidi): dunque nella maggior parte dei casi questa metodologia non è applicabile. Per le proteine di dimensioni medie si deve invece ricorrere a metodi che si basano sul confronto della struttura da determinare con quelle di proteine con strutture già determinate con metodi di indagine strutturale. In ogni caso, qualsiasi sia il modo con cui una struttura proteica viene predetta per via computazionale, è indispensabile sottoporla a una successiva validazione: vale a dire, bisogna ottenere evidenze sperimentali che confermino la correttezza della predizione. Per esempio, se si scopre che un dato amminoacido ha un ruolo importante nella stabilità della molecola, usando metodiche di biologia molecolare bisognerà sostituirlo con un altro che per le sue proprietà non può avere lo stesso ruolo, e valutare se la proteina ha perso stabilità a seguito della sostituzione.
Sulla base di queste considerazioni si potrebbe pensare che la natura delle difficoltà riscontrate nei problemi sperimentali menzionati non sia qualitativamente diversa da quella incontrata nei casi, menzionati in precedenza, delle cinetiche chimiche e della predizione della saturazione dell’emoglobina. In altre parole, potrebbe trattarsi soltanto della necessità di sviluppare modelli matematici molto più complessi. Senza dubbio è vero che la difficoltà è anche legata allo strumento matematico, talché già al livello di complessità della struttura delle proteine (peraltro ben più elementare di quello, per esempio, di una cellula intera), è impossibile in assoluto ricavare equazioni che descrivano in modo analitico una data proprietà del sistema, come appunto la struttura stessa. Le equazioni praticamente utilizzabili devono infatti basarsi inevitabilmente su una serie di semplificazioni.
Ciò nondimeno, il problema della predizione della funzione di una proteina a partire dalla struttura mette in evidenza con chiarezza che non si tratta solo di questo. Innanzitutto, come dato di fatto tali tentativi non vengono attuati adottando procedure a priori, cioè che siano ristrette allo studio della molecola in sé, ma confrontando sequenza e struttura di una proteina a funzione ignota con sequenza e struttura di proteine a funzione nota. Se tra le due proteine si riscontrano somiglianze strutturali e di sequenza significative, è anche probabile che esse abbiano la stessa funzione. Ma il punto più sostanziale che mette in evidenza l’irriducibilità delle proprietà emergenti a un certo livello a quelle di livello inferiore, risiede nel fatto che è intrinsecamente impossibile affrontare il problema del ruolo biologico di una proteina a prescindere dal contesto in cui essa si trova: certe proprietà, come per esempio la sensibilità a determinate molecole che la attivano o inibiscono, si comprendono solo in relazione alla funzione che essa esercita nell’ambiente in cui si trova.
In altre parole, se anche fosse possibile dedurre da sequenza e struttura di una data proteina che essa è sensibile a certi composti, tale osservazione nulla ci direbbe in merito al significato funzionale di un tale fenomeno, che è poi un aspetto centrale in ordine all’interpretazione del ruolo biologico della molecola.
La plurilivellarità: una connotazione ineludibile della logica costruttiva dei sistemi biologici
Molto di quanto abbiamo esposto nel paragrafo precedente si riduce a un concetto ben noto al biologo che va sotto il nome di plurilivellarità, e che si può così definire: le proprietà biologiche che si riscontrano a un certo livello di complessità di un sistema non sono semplicemente deducibili da quelle che governano il sistema ai livelli inferiori.
Oltre al caso menzionato delle proteine, un altro esempio che in modo altrettanto significativo documenta questo concetto è quello degli acidi nucleici, depositari dell’informazione genetica. Con la sua sequenza di basi, un tratto di DNA codifica di norma la sequenza amminoacidica di una proteina. Eppure lo studio di una molecola di acido nucleico con metodiche puramente chimiche (determinandone, per esempio, la sua polarità, il peso molecolare, le proprietà ottiche, ma anche la struttura tridimensionale complessiva) non ci direbbe nulla in relazione alla sua proprietà essenziale, che è lo specifico contenuto informativo, decifrabile solo in base al codice genetico, evidentemente una proprietà estrinseca alle molecole di DNA.
In sintesi, le proprietà emergenti delle molecole biologiche obbediscono senza dubbio alle leggi che governano i livelli inferiori di complessità, come le normali leggi della fisica, della chimica generale e organica, ma sono di più ed altro di queste stesse leggi. Ciò non toglie evidentemente che per la comprensione delle proprietà emergenti delle molecole biologiche sia indispensabile una conoscenza approfondita delle proprietà chimico-fisiche del livello inferiore, perché queste determinano le condizioni al contorno, alle quali le molecole biologiche debbono comunque conformarsi.
Gli sviluppi recenti dell’approccio plurilivellare: la biologia dei sistemi
Una recente pubblicazione scientifica delinea con chiarezza i tratti di una nuova disciplina, la biologia dei sistemi, che rappresenta un nuovo modo di vedere, e conseguentemente di investigare i sistemi biologici: «Lo studio delle funzioni degli organismi viventi non può essere impostato adeguatamente occupandosi delle molecole una per volta, anche se venissero studiate tutte. Tali studi non chiarirebbero le proprietà funzionali sopramolecolari come il ciclo cellulare, gli stati metabolici e le (dis)funzioni cellulari. Non consentirebbero di comprendere le malattie multifattoriali […]. Per comprendere funzioni e disfunzioni degli organismi è necessario un approccio di sistema […]. I successi delle scienze biomolecolari hanno portato alla caratterizzazione della maggior parte dei componenti molecolari di molti organismi. Questo successo ha fatto spostare l’attenzione della ricerca biologica dalle singole molecole alle reti.» [Bruggeman & Westerhoff (2007) Trends Microbiol 15, 45-50].
Dunque la biologia dei sistemi non investiga un sistema biologico pezzo per pezzo, come tradizionalmente si era soliti fare, ma considerandolo nel suo insieme e valutando le proprietà complessive che emergono dal sistema stesso. Questa rivoluzione scientifica è stata resa possibile da sostanziali progressi tecnologici in vari campi, in primis quello del sequenziamento genico, della spettroscopia di massa, delle metodiche di identificazione ad alta resa delle proteine, dell’informatica. Si sono sviluppati così dei campi di indagine (le omiche) che attuano questi approcci complessivi verso diverse classi di composti biologici, come illustrato nel riquadro qui di seguito.
| Genomica | L’insieme dei geni di un organismo |
| Proteomica | L’insieme delle proteine di un organismo |
| Interattomica | L’insieme delle interazioni tra le proteine di un organismo |
| Metabolomica | L’insieme degli intermedi metabolici di un organismo |
L’interattomica è probabilmente l’approccio che meglio può documentare la logica della biologia dei sistemi. Nell’immagine seguente viene presentata una parte dell’interattoma dei neuroni dell’ippocampo (in alto). In diversi stati patologici si osserva l’attivazione di una parte della rete complessiva di interazioni (evidenziato in basso). La rete si può considerare come un sistema integrato che risponde a un certo input (stato patologico, farmaco) producendo un output definito. La speranza è di poter predire in un futuro (probabilmente non molto prossimo) cosa un certo input produca in termini di output (risposta del sistema biologico). Sarebbe così possibile comprendere il meccanismo di azione di un farmaco, oppure la migliore proteina bersaglio per un trattamento farmacologico.
La rete si può considerare come un sistema integrato che risponde a un certo input (stato patologico, farmaco) producendo un output definito. La speranza è di poter predire in un futuro (probabilmente non molto prossimo) cosa un certo input produca in termini di output (risposta del sistema biologico). Sarebbe così possibile comprendere il meccanismo di azione di un farmaco, oppure la migliore proteina bersaglio per un trattamento farmacologico.
In un sistema biologico esiste una specifica componente a cui sia riconducibile la peculiarità della vita?
Gli esempi che abbiamo prodotto mostrano come complessità e plurilivellarità siano prerogative di tutti i sistemi viventi. La domanda che può emergere, sulla scorta di queste considerazioni, è se sia possibile identificare qualche elemento molecolare che preminentemente rispetto ad altri sia quello caratterizzante il fenomeno vita.
Per rispondere a questa domanda è opportuno innanzitutto introdurre un aspetto metodologico: qualsiasi intervento di uno sperimentatore su un sistema biologico a scopo di studio comporta inevitabilmente di perturbarlo in qualche modo. Nel caso più tipico, per poter investigare le proprietà dei componenti cellulari bisogna separarli l’uno dall’altro, distruggendo così l’integrità strutturale della cellula. Tale procedura si chiama frazionamento subcellulare (illustrato schematicamente nell’immagine seguente). Ciò pone innanzitutto un problema di metodo, in quanto lo studio dei «pezzi» individuali derivanti dalla frammentazione di una cellula (gli organuli cellulari), pone l’interrogativo di come essi effettivamente operino in vivo, cioè quando sono normalmente funzionanti perché integrati nel sistema cellulare. A questo riguardo, il biochimico David Green scriveva ironicamente che un ingegnere in gamba potrebbe costruire un aspirapolvere dai pezzi di un’auto, ma ciò non significa che le auto contengano aspirapolveri! Si pone quindi il problema di una corretta interpretazione del dato ottenuto con questa procedura sperimentale, al fine di comprendere l’effettiva modalità di funzionamento delle parti nel loro contesto naturale. Tutto ciò riecheggia quanto avevamo menzionato in precedenza in relazione alla comprensione del ruolo biologico delle proteine.
Ciò pone innanzitutto un problema di metodo, in quanto lo studio dei «pezzi» individuali derivanti dalla frammentazione di una cellula (gli organuli cellulari), pone l’interrogativo di come essi effettivamente operino in vivo, cioè quando sono normalmente funzionanti perché integrati nel sistema cellulare. A questo riguardo, il biochimico David Green scriveva ironicamente che un ingegnere in gamba potrebbe costruire un aspirapolvere dai pezzi di un’auto, ma ciò non significa che le auto contengano aspirapolveri! Si pone quindi il problema di una corretta interpretazione del dato ottenuto con questa procedura sperimentale, al fine di comprendere l’effettiva modalità di funzionamento delle parti nel loro contesto naturale. Tutto ciò riecheggia quanto avevamo menzionato in precedenza in relazione alla comprensione del ruolo biologico delle proteine.
Ma esiste un aspetto ancora più profondo legato a questa specifica problematica: scomponendo un sistema biologico nei suoi componenti, l’aspetto più proprio della vita non può essere identificato in nessuno di questi. Tale concetto è stato enunciato magistralmente dal biologo molecolare Erwin Chargaff: «L’analisi delle parti che compongono un organismo vivente comporta, salvo poche eccezioni, il venir meno dell’elemento essenziale della vita stessa […]. L’insufficienza delle scienze nei confronti della vita ha ragioni profonde. Probabilmente non è un caso che tra tutte le scienze sia proprio la biologia quella che non riesce a definire l’oggetto che studia: noi non disponiamo di una definizione scientifica della vita. In effetti sono solo cellule e tessuti morti quelli che vengono sottoposti alle analisi più dettagliate.» [E. Chargaff (1980), Mistero impenetrabile]
In sintesi, ciò che caratterizza il sistema biologico è la sua «irriducibilità»: questo breve excursus rende evidente che esso sempre è di più e altro che non la sommatoria delle singole componenti.
Paolo Tortora
(Professore ordinario di Biochimica, Dipartimento di Biotecnologie e Bioscienze, Università degli Studi di Milano Bicocca)
© Pubblicato sul n° 43 di Emmeciquadro