
 “for the development of multiscale models for complex chimica systems”
“for the development of multiscale models for complex chimica systems”
Il Premio Nobel per la Chimica 2013 è stato attribuito a Martin Karplus, Michael Levitt e Arieh Warshel, per il loro fondamentale contributo allo sviluppo dei metodi computazionali che oggi permettono lo studio di sistemi chimici complessi.
Per comprendere appieno il significato delle ricerche che hanno portato all’assegnazione del premio Nobel per la chimica 2013 è opportuno fare una premessa. Negli ultimi cinquanta anni la chimica e la biochimica si sono sviluppate e trasformate in modo estremamente rapido. E questo rapido sviluppo, così come il modo in cui queste discipline si sono trasformate, è ben esemplificato da ciò che e’ accaduto nell’ambito della chimica delle biomolecole. Mentre da un lato le tecniche spettroscopiche, quali la diffrazione di raggi X e la risonanza magnetica nucleare (NMR), hanno assunto un ruolo sempre più centrale nelle ricerche finalizzate a svelare la struttura tridimensionale di sistemi molecolari complessi quali enzimi, recettori, DNA e RNA, il concomitante sviluppo e utilizzo di metodi computazionali basati sulla meccanica classica e/o sulla meccanica quantistica, come quelli sviluppati da Martin Karplus, Michael Levitt e Arieh Warshel, ha permesso di complementare le informazioni ottenute per via sperimentale, permettendo una sempre più approfondita indagine e comprensione delle proprietà strutturali, dinamiche ed elettroniche di sistemi chimici estremamente complessi. Ma la conoscenza delle proprietà strutturali di sistemi molecolari complessi quali le proteine è solo il primo passo verso la comprensione delle relazioni che legano la struttura di questi sistemi molecolari alla loro funzione. E infatti, negli ultimi anni l’attenzione dei ricercatori si è focalizzata principalmente sullo studio degli aspetti funzionali dei sistemi molecolari complessi di origine naturale.
La conoscenza delle strutture tridimensionali di molte biomolecole complesse è stata quindi la piattaforma su cui si sono fondati tutti quegli studi indirizzati a comprendere le basi molecolari della loro attività biologica. Infatti, i sistemi molecolari complessi di rilevanza biologica hanno una peculiarità che li differenzia dalle altre molecole che possiamo osservare in natura.
Alla struttura tridimensionale delle biomolecole complesse (quali ad esempio le proteine) è sempre associata una funzione specifica. Di conseguenza uno degli obiettivi centrali della chimica e delle biochimica degli ultimi decenni è stato quello di svelare le relazioni esistenti tra la struttura di questi complessi sistemi molecolari naturali e la loro funzione. In altre parole, le biomolecole complesse si possono descrivere come dei veri e propri dispositivi molecolari, la cui funzione dipende dalle loro caratteristiche strutturali. In particolare, le proteine sono sistemi molecolari che sono stati «plasmati», generazione dopo generazione, dalla selezione naturale, cioè da un processo che prevede l’introduzione di modificazioni casuali nella sequenza amminoacidica della proteina e la selezione delle varianti con le proprietà funzionali più adeguate per un efficace adattamento all’ambiente circostante.
È opportuno ricordare che, parallelamente a questo tipo di ricerche riguardanti i sistemi chimici complessi di origine naturale, i chimici sono ovviamente anche coinvolti nella progettazione di dispositivi molecolari di natura sintetica caratterizzati da una specifica funzione (si pensi per esempio ai dispositivi molecolari usati nelle nanotecnologie e nella scienza dei materiali). Ma mentre nel classico processo di progettazione di sistemi molecolari complessi è il chimico a «disegnare» e sintetizzare il dispositivo molecolare, in modo che questo possa svolgere una ben determinata funzione, nel caso delle ricerche riguardanti i dispositivi molecolari di origine naturale (e quindi non progettati dall’uomo) i ricercatori devono prima di tutto rispondere alla domanda «quale è la struttura?» (che è requisito imprescindibile per comprendere la funzione), ma poi cercare di ottenere risposte alla domanda «quale è il meccanismo con cui questi dispositivi molecolari complessi svolgono in modo così efficiente la loro funzione?».
Questo modo di affrontare la ricerca può essere considerato come un vero e proprio processo di ingegneria inversa, dove lo scopo è quello di comprendere i dettagli associati al funzionamento di un dispositivo, conoscendone la struttura, ma non potendo consultare nessun «libretto di istruzioni».
In entrambi gli scenari (progettazione di molecole complesse e ingegneria inversa di biomolecole), tecniche sperimentali come la marcatura isotopica e le spettroscopie con risoluzione dell’ordine dei femtosecondi, possono fornire risultati fondamentali per chiarire le relazioni tra struttura e attività, ma raramente è possibile rivelare in modo completo il meccanismo di funzionamento di sistemi molecolari complessi, e quindi svelare le relazione tra la struttura e la funzione, utilizzando solamente queste metodologie. Conseguentemente, i metodi computazionali adatti a studiare i sistemi molecolari complessi hanno rappresentato, e sempre più rappresentano, degli strumenti fondamentali per complementare gli studi effettuati utilizzando tecniche sperimentali.
Struttura degli enzimi e loro attività
Per discutere un esempio specifico, particolarmente rilevante nelle biotecnologie, vediamo come i metodi computazionali sviluppati dai tre premi Nobel per la chimica 2013 possono permettere di svelare le relazioni esistenti tra la struttura degli enzimi e la loro attività.
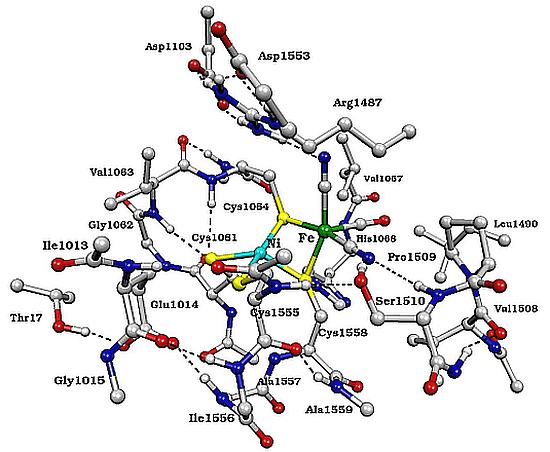 Struttura del sito attivo dell’enzima [NiFe]-idrogenasi
Struttura del sito attivo dell’enzima [NiFe]-idrogenasi
Questo enzima, che catalizza l’ossidazione reversibile dell’idrogeno molecolare, e di conseguenza ha delle potenziali applicazioni in ambito energetico e biotecnologico, è stato oggetto di diversi studi mirati a chiarire il meccanismo con cui l’enzima catalizza tale reazione. Molti di questi studi sono stati effettuati utilizzando le più recenti varianti dei metodi computazionali sviluppati da Martin Karplus, Michael Levitt e Arieh Warshel.
Gli enzimi sono proteine che riescono a catalizzare delle specifiche reazioni (sono cioè sostanze che, aggiunte in piccole quantità, aumentano la velocità con cui avviene una reazione, senza venir consumate durante la reazione stessa). Questo incremento nella velocità della reazione dipende dal fatto che il sito attivo dell’enzima, dove avviene la trasformazione dei reagenti della reazione nei corrispondenti prodotti, ha delle caratteristiche chimico-fisiche tali da permettere alla reazione di avvenire secondo un meccanismo differente, e che prevede il superamento di barriere energetiche più basse, rispetto alla analoga trasformazione in assenza dell’enzima. Questo risulta in un abbassamento della cosiddetta energia di attivazione del processo e di conseguenza in un aumento (generalmente di diversi ordini di grandezza) della velocità con cui la trasformazione avviene.
Per capire come il sito attivo riesce ad abbassare la barriera energetica che connette i reagenti e i prodotti della reazione, e’ fondamentale ottenere informazioni su quello che in gergo scientifico viene definito lo stato di transizione della reazione, cioè la disposizione degli atomi della molecola mentre si sta trasformando da reagenti a prodotti (e quindi è in uno stato instabile a cui corrisponde una energia più elevata che nei reagenti e nei prodotti).
Tali informazioni si possono ottenere proprio con i metodi computazionali sviluppati da Martin Karplus, Michael Levitt e Arieh Warshel, in quanto mediante tali metodi è possibile studiare il meccanismo con cui avviene una reazione, calcolando energie e strutture di intermedi e stati di transizione. In particolare, la struttura dello stato di transizione e la sua energia relativa rispetto a reagenti e prodotti, sono informazioni fondamentali per il chimico e il biochimico che vogliono comprendere a fondo come e perché un particolare enzima riesce a rendere una reazione così veloce.
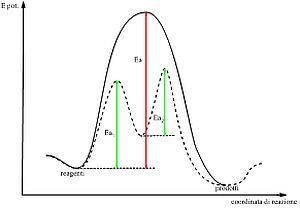 [A sinistra: Profilo di reazione per una generica reazione chimica]
[A sinistra: Profilo di reazione per una generica reazione chimica]
Tali informazioni non sono importanti solo per la comprensione dei meccanismi molecolari alla base della catalisi, ma possono permettere la «progettazione» di varianti dell’enzima o di composti di sintesi con specifiche proprietà catalitiche, e quindi di elevato interesse applicativo in diversi ambiti.
Nell’immagine la curva continua rappresenta l’andamento dell’energia in funzione della coordinata di reazione (quindi in funzione del meccanismo molecolare con cui i reagenti si trasformano in prodotti) per la reazione non catalizzata. Il corrispondente valore di energia di attivazione (Ea), e quindi della barriera energetica da superare perché la reazione avvenga, è mostrato in rosso. La curva tratteggiata rappresenta il grafico per la reazione catalizzata, con le corrispondenti energie di attivazione in verde.
Punti di forza dei metodi computazionali
Una volta compreso il significato e l’applicabilità dei metodi sviluppati dai tre premi Nobel per la chimica 2013, entriamo nel dettaglio dei principi su cui tali metodi si fondano. Come detto, il riconoscimento è stato attribuito per gli studi che hanno portato allo sviluppo di metodi computazionali basati sia sulla meccanica classica che sulla meccanica quantistica.
Quando una molecola viene descritta utilizzando la meccanica quantistica, gli elettroni della molecola vengono considerati esplicitamente. Il pregio di questo tipo di rappresentazione è che tutte le proprietà direttamente dipendenti dalla struttura elettronica possono essere calcolate in modo accurato (strutture di intermedi di reazione e stati di transizione, energie di attivazione, proprietà magnetiche, proprietà spettroscopiche, eccetera).
Di contro, i calcoli necessari per questo tipo di descrizione sono molto onerosi, in quanto per descrivere adeguatamente gli elettroni è necessario effettuare calcoli complessi che richiedono tempi molto lunghi, e quindi tali metodi non permettono ancora di indagare sistemi complessi come intere proteine. Al contrario, nei sistemi molecolari, o nelle porzioni di questi, che vengono descritti utilizzando una trattazione fisica di tipo classico (a cui ci si riferisce solitamente con il termine Meccanica Molecolare), gli elettroni non sono descritti esplicitamente, e la loro presenza è tenuta solo implicitamente in considerazione (per esempio attribuendo una carica elettrica ai diversi atomi della molecola, sulla base della loro elettronegatività, in modo che l’attrazione e repulsione elettrostatica tra atomi diversi sia descritta in modo sufficientemente accurato).
Di conseguenza solo alcune proprietà molecolari possono essere descritte adeguatamente (tutte quelle che non dipendono esplicitamente dagli elettroni), ma i calcoli diventano molto meno onerosi e quindi si possono indagare le proprietà di sistemi molecolari molto complessi come proteine, acido nucleici e membrane biologiche. Uno dei punti di forza dei metodi computazionali sviluppati da Martin Karplus, Michael Levitt e Arieh Warshel è proprio quello di permettere l’utilizzo simultaneo di una descrizione classica e quantistica per descrivere diverse porzioni del sistema molecolare oggetto di studio, e quindi rendere possibile lo studio per via computazionale di sistemi complicati come per esempio gli enzimi (che possono essere formati da centinaia di migliaia di atomi).
La possibilità di utilizzare questi metodi ibridi (solitamente definiti come modelli QM/MM, in quanto una porzione della molecola è descritta mediante la Quanto-Meccanica, mentre tutto il resto del sistema molecolare è descritto mediante la Meccanica Molecolare) offre molti vantaggi e permette di studiare proprietà e fenomeni che sarebbero altrimenti difficilmente indagabili con altre tecniche.
In particolare, la regione del sistema oggetto dello studio dove avvengono le trasformazioni chimiche che implicano scissione o formazione di legami covalenti, o il trasferimento di elettroni da un punto a un altro della molecola, viene descritta usando la meccanica quantistica.
Ma a causa dei tempi proibitivi che sarebbero necessari per studiare un sistema molecolare complesso usando una descrizione completamente quantistica, nei metodi sviluppati dai tre vincitori del premio Nobel 2013 le porzioni del sistema dove non avvengono fenomeni direttamente dipendenti dalla presenza degli elettroni vengono descritte utilizzando una descrizione fisica più semplificata, ma che permette comunque di descrivere adeguatamente tutta una serie di fenomeni molecolari importantissimi (e che possono influenzare le proprietà della porzione molecolare descritta con la meccanica quantistica), quali per esempio le modificazioni conformazionali che avvengono nelle proteine a seguito dell’interazione con un’altra molecola. Inoltre, in questi modelli computazionali ibridi la porzioni della molecola trattate con la fisica classica e quantistica interagiscono tra di loro in maniera fisicamente plausibile, e quindi si influenzano reciprocamente, rendendo il modello computazionale sufficientemente accurato per studiare una ampia gamma di fenomeni.
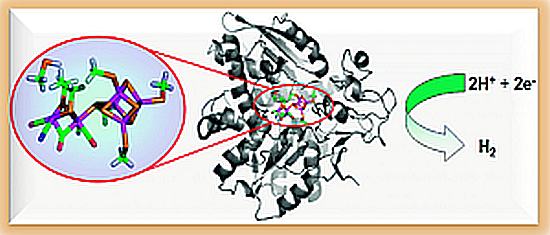 Rappresentazione schematica della struttura dell’enzima [FeFe]-idrogenasi
Rappresentazione schematica della struttura dell’enzima [FeFe]-idrogenasi
L’immagine mostra la rappresentazione schematica della struttura dell’enzima [FeFe]-idrogenasi, che catalizza l’ossidazione reversibile dell’idrogeno molecolare, e ha potenziali applicazioni in ambito energetico e biotecnologico.
Per comprendere il meccanismo con cui l’enzima catalizza tale reazione sono stati effettuati, in anni recenti, diversi studi computazionali basati su modelli QM/MM, dove il sito attivo dell’enzima è stato descritto mediante una descrizione quantomeccanica per studiarne la reattività con protoni ed elettroni, mentre gli effetti dell’intorno proteico sulla reattività del sito attivo sono stati modellati in modo più semplificato utilizzando una descrizione classica.
Il contributo dei tre premi Nobel – breve analisi storica Il primo passo nello sviluppo di modelli computazionali ibridi è stato fatto negli anni settanta, quando Arieh Warshel trascorse un periodo nei laboratori di Martin Karplus, ad Harvard, per studiare le proprietà delle molecole che giocano un ruolo fondamentale nei meccanismi molecolari associati alla visione nei mammiferi, come il retinale. A tale scopo, i due ricercatori svilupparono il primo programma che permettesse di descrivere diverse porzioni delle molecole di interesse usando una diversa descrizione fisica del sistema. Nel caso specifico, i nuclei e gli elettroni sigma vennero modellati usando un approccio derivante dalla fisica classica, mentre gli elettroni pi-greco vennero rappresentati mediante una descrizione quantomeccanica. Poco tempo dopo Arieh Warshel e Michel Levitt generalizzarono tale approccio, proponendo anche diverse strategie per affrontare uno degli aspetti più critici dei modelli ibridi, cioè la descrizione più opportuna dell’interazione tra le parti del sistema modellate mediante una descrizione quantistica e classica. Negli stessi anni i due scienziati posero le basi per studiare sistemi di dimensioni sempre maggiori (e che quindi potessero permettere l’indagine di sistemi molecolari di grande interesse come proteine e acidi nucleici), mostrando che era possibile descrivere interi gruppi di atomi come un’unica unità rigida (i cosiddetti pseudo-atomi), e di conseguenza rendere i calcoli meno onerosi. |
Prospettive future
Gli studi dei tre premi Nobel per la chimica 2013 sono stati la base su cui si è fondato, in anni più recenti, lo sviluppo di metodi ibridi sempre più accurati, e la loro applicazione in svariati settori che spaziano dalla chimica organica e inorganica, alla biochimica, fino ad arrivare allo studio dei meccanismi molecolari in processi catalitici in fase eterogenea e al calcolo delle proprietà spettroscopiche di sistemi complessi in soluzione.
Tra i ricercatori più attivi in questo campo vanno citati J. Gao, F. Maseras, K. Morokuma, P. Kollman, W. Thiel e U. Ryde. Ma è forse ancora più importante sottolineare come lo sviluppo di questi metodi abbia stimolato una molto fruttuosa collaborazione tra gruppi di ricerca sperimentali e computazionali, permettendo di affrontare e risolvere problematiche chimiche di grande rilevanza in ambito biologico e medico, nella catalisi, e nella progettazione di nuovi materiali.
Vai all’articolo in formato PDF
Luca De Gioia
(Professore Associato di Chimica Generale e Inorganica presso il Dipartimento di Biotecnologie e Bioscienze dell’Università degli Studi di Milano – Bicocca)
© Pubblicato sul n° 51 di Emmeciquadro